力学所在固态储氢材料研究方面取得新进展
化石能源对环境的污染破坏使得社会发展不可持续,人们正积极努力寻找新一代的绿色能源。在这过程中,氢能因其丰富的储量、环境友好的特性、高能量密度以及其产生的唯一产物——水,而备受瞩目。虽然氢气生产的途径多种多样,但是氢气的储存仍然是一个极大的挑战。传统的高压氢气储存方法要求持续维持高压,由此带来的潜在风险不容忽视。另一方面,液态储氢虽然是一种可行的方法,但其实现所需的能源投入较大,且效率相对较低。因此,固态氢气储存被认为是一种理想的解决方案。固态储存能够在不改变材料结构的前提下,吸收和释放大量的氢气。目前的关键问题在于寻找一种储氢性能出色且能在吻合条件下释放氢气的固态储氢材料。美国能源部提出了关于固态氢气储存的指导方针。根据这些方针,一个有效的固态储氢装置应当能够储存至少6.5 wt%的氢气,以满足氢燃料电池车续航500英里(约805公里)的目标。为了实现这一目标,研究人员们正在不遗余力地探索各种可能的固态储氢材料,以在储存和释放氢气时均能表现出优异的性能。
中国科学院力学所的彭庆研究员与广西大学的欧阳义芳教授领导的团队,通过第一性原理计算,成功开发出一种具有广阔前景的二维固态储氢材料体系Ti-decorated Irida-Graphene(钛修饰的鸢尾花型石墨烯,简称TIG)。这一体系中,Irida-Graphene(鸢尾花型石墨烯,简称IG)是一种新型的类石墨烯材料,由三原子、六原子和八原子的碳环组成。研究团队发现,通过在IG上引入修饰性的钛(Ti)原子,其储氢性能可以高达7.7wt%。
首先,研究团队对钛原子在鸢尾花型石墨烯不同吸附位点的吸附能力进行了计算研究,发现鸢尾花型石墨烯的六原子上方的中空位(Hollow)是最稳定的吸附位点。随后,通过在体系中逐个加入氢分子(H2),计算了连续吸氢能力,结果显示每个钛原子周围可吸附5个氢分子。因为鸢尾花型石墨烯是二维材料具有上下两个表面,其两个表面可以同时吸附氢分子。当钛原子填满鸢尾花型石墨烯的所有六原子环中心环位时,其可储存的氢气高达7.7 wt%,超过了美国能源部提出的6.5 wt%目标。根据6.5 wt%的储氢密度可以使氢燃料电池车续航500英里估计,7.7 wt%储氢密度的续航约为590英里(950公里)
应用Van't Hoff方程计算得知,TIG的平均放氢温度为524 K(253摄氏度)。电子结构分析结果表明,氢气分子与TIG之间为Kubas型键合。氢气分子在吸附后获得净电荷。这种电荷转移还会导致H-H键距延长,但不会使分子与其结构分离。同时在TIG储存氢气时,钛原子的3d轨道与纯TIG相比,接近费米能级的电子态密度有所减少,表明在吸附后电荷从钛原子的3d轨道转移到氢分子的1s轨道上。随着额外的氢气分子不断集成到系统中,钛原子的3d轨道上电子将继续转移到氢的1s轨道上,形成稳定吸附。
为了考量TIG材料的结构热稳定性,研究团队还通过CI-NEB方法探究了修饰的钛原子的迁移能力,发现钛原子的扩散迁移能垒为5.0 eV。在平均解吸温度524 K下,钛原子的热能为0.68 eV,远低于扩散能垒值。由于结构构型的稳定性以及高能垒值在反应温度下的限制,钛原子的迁移受到限制。这意味着在吸氢和放氢过程中,钛原子不会发生迁移,从而避免了金属团聚引发的储氢结构破坏,证明了该体系作为可行的储氢介质。最后,研究团队还借助第一性原理分子动力学模拟确定了该材料在室温300 K和高温600 K下的热力学稳定性。所有这些理论预测结果为开发新型高效的储氢材料提供了一种新的可选方案。
该研究成果以“Stable and 7.7 wt% hydrogen storage capacity of Ti decorated Irida-Graphene from first-principles calculations”为题发表于International Journal of Hydrogen Energy期刊上。广西大学博士生谭咏康为论文第一作者,彭庆研究员和欧阳义芳教授为通讯作者。此工作得到国家自然科学基金和中国科学院力学所力英计划的支持。
论文链接:
https://doi.org/10.1016/j.ijhydene.2023.08.115
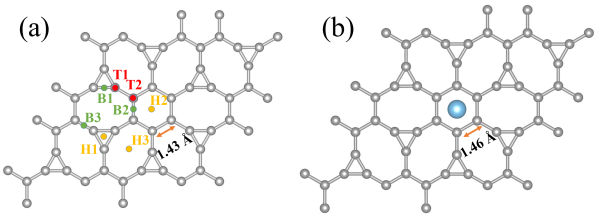
图 1. (a)2×2 的IG超胞;(b)TIG的原子结构。其中钛原子位于H2中空位点的吸附能最低,结构最稳定。灰色和蓝色球体分别表示碳原子和钛原子。黄色、红色和绿色分别表示中空位、顶位和桥位钛原子吸附位点。通过计算结合能发现,钛原子可以吸附在中空位和顶位,结合能分别为T1 (-2.50 eV)、T2 (-2.17 eV)、H1(-2.33 eV)、H2(-2.84 eV)和H3(-2.77 eV)。其种Ti原子在六角碳环的中空位的结合能最低。在六角碳环上方装饰了钛原子后,六角碳环的C-C键从1.43 ![]() 增加到1.46
增加到1.46 ![]()
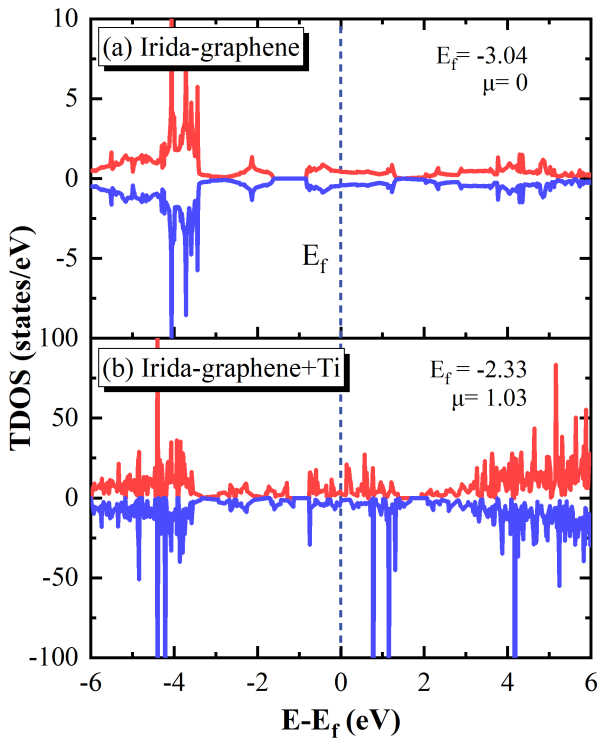
图 2 IG在钛原子修饰之前(a)和之后(b)的总态密度(TDOS)。其中费米级 Ef被设为零。在IG中掺入钛原子后,可能态的密度得到了提高,自旋向上和自旋向下的态密度的对称性消失,且系统变得具有磁性
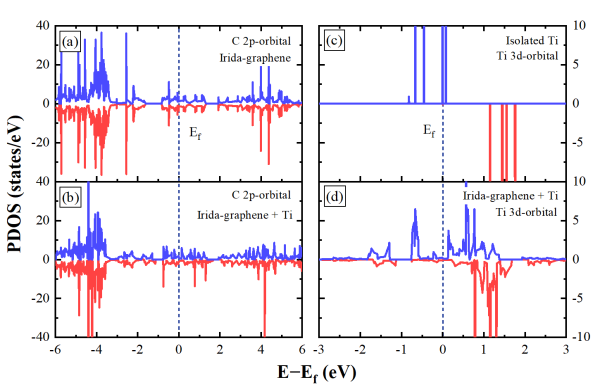
图 3 碳原子的2p轨道在(a)IG和(b)TIG的投影态密度(PDOS);钛原子3d轨道在(c)孤立状态和(d)TIG中的投影状态密度。随着体系中钛原子掺入后,碳的2p轨道在费米能级以下的状态数显著增加,说明电荷增加。同时,钛原子的3d轨道从离散态变为连续态,表明钛原子与IG之间发生结合
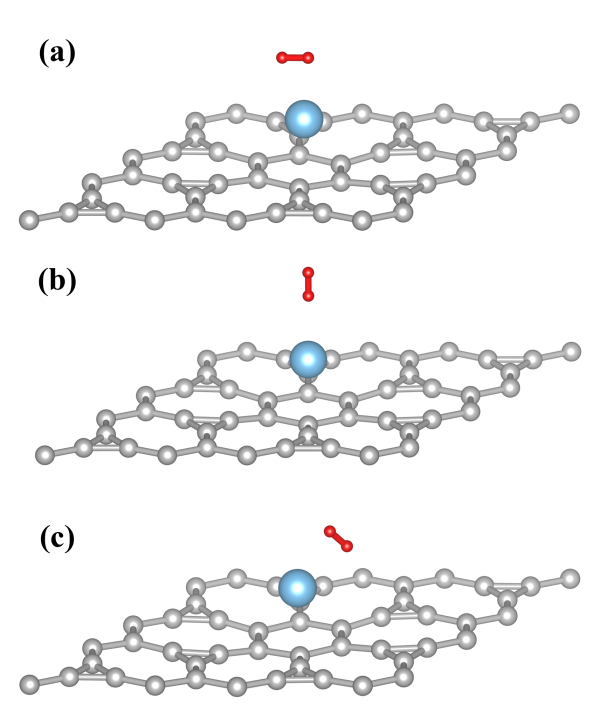
图 4 氢分子以不同方向放置于钛原子周围,并经过几何优化后的原子结构。(a)水平方向、(b)垂直方向和(c)倾斜方向。这些不同方向放置的氢分子的吸附能分别为-0.40、-0.13和-0.60 eV。由此可见倾斜放置是最稳定的吸附状态
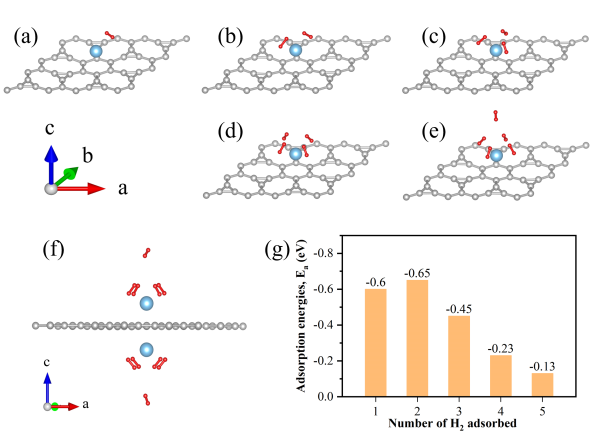
图 5 TIG吸附H2分子后的优化结构和吸附能。(a)~(e) 1~5 H2分子吸附结构。(f) 在IG两侧均放置钛原子,每个钛原子吸附5个H2的结构。灰球、蓝球和红球分别代表碳、钛和氢原子。(g) H2连续吸附能
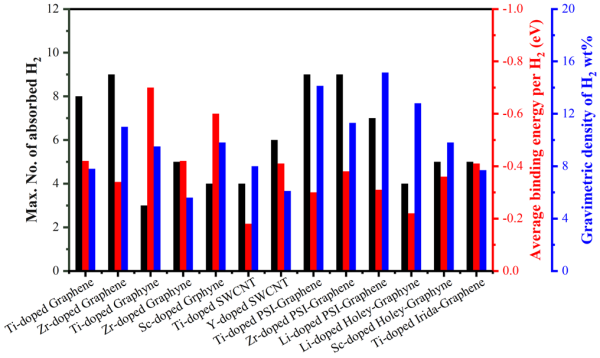
图 6 各种金属修饰碳纳米材料的储氢性能。可以观察到TIG的体系具有相对较高的储氢效率和合适的解吸温度。这表明用TIG是一种有价值的储氢潜在材料
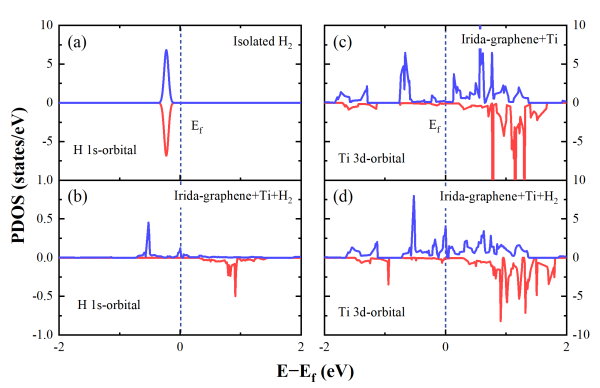
图 7 氢原子的1s轨道和钛原子3d轨道的投影态密度(PDOS)。(a)孤立H2的氢1s轨道;(b)TIG+H2系统的氢1s轨道;(c)TIG的钛3d轨道;(d)TIG+H2系统的钛3d轨道。系统吸收H2后,氢的1s轨道在费米级附近的密度增加。钛的3d轨道则在费米水平以下下降。说明吸氢后,钛原子的3d轨道的电荷向氢的1s轨道转移
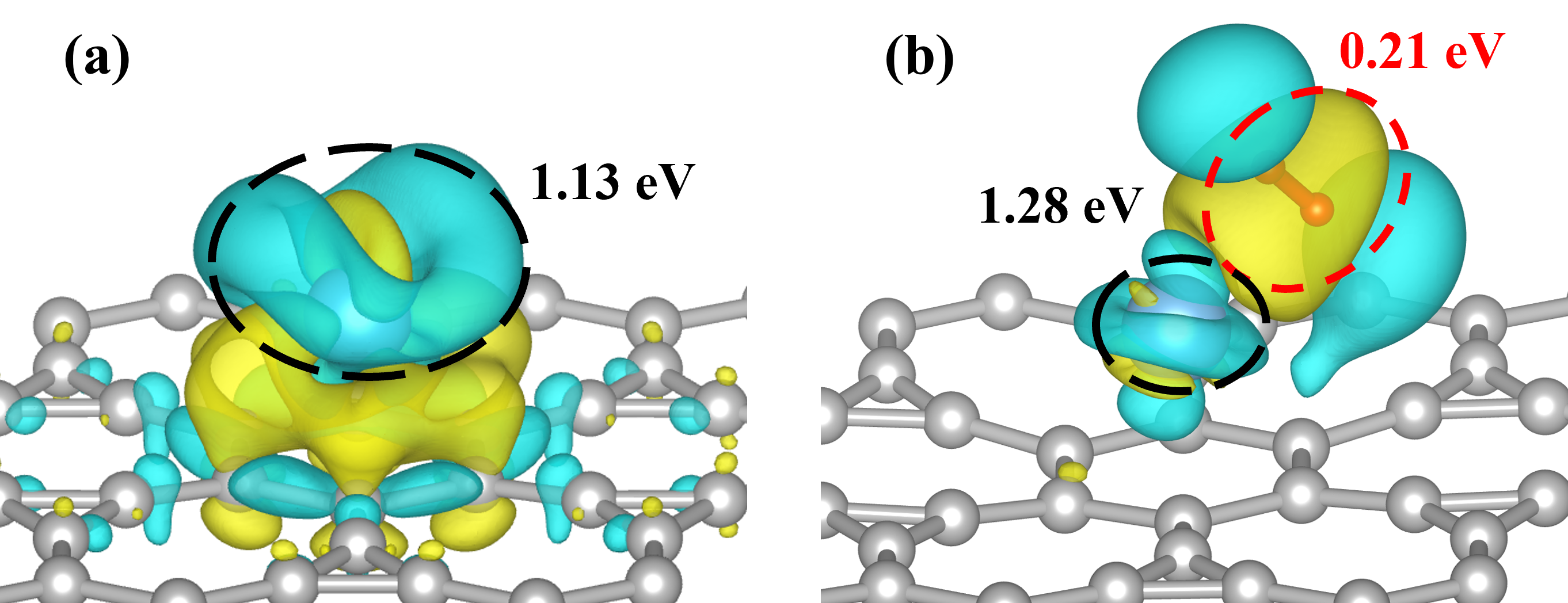
图 8 TIG体系(a)在吸附第一个氢分子之前和(b)在吸附第一个氢分子之后的差分电荷密度。电荷密度图中黄色代表静电势浓度增加,蓝色代表浓度降低。根据Bader电荷分析,TIG中钛原子将1.13e的电荷转移到相邻的碳原子上,这是钛原子和IG之间形成强键的原因
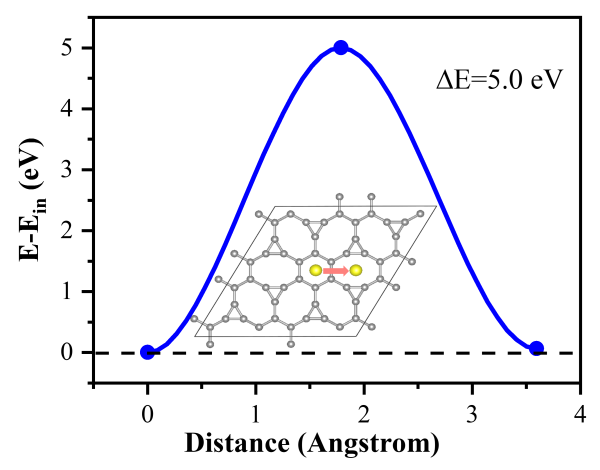
图9钛原子从六角中空位向周围的八角环中空位迁移扩散能垒。插图显示了扩散路径。初始状态的能量设定为0 eV,最终状态比初始状态高0.07 eV。在此扩散路径上的扩散能垒为5.0 eV。这保证了钛原子在服役条件下发生迁移而导致的金属聚集和结构破坏
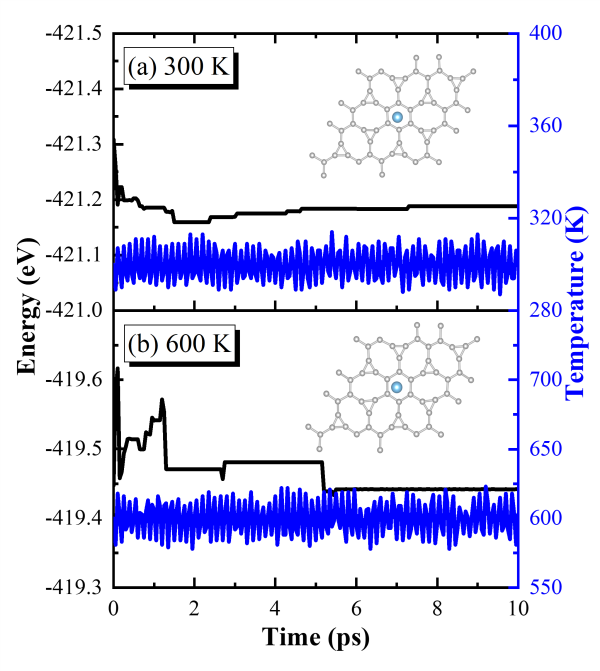
图 10 第一性原理分子动力学模拟在NVT系综下TIG的总能量和温度随模拟时间变化的关系曲线。模拟温度分别为(a)300 K和(b)600 K。(a)和(b)中的插图分别是TIG在两个温度下热浴10 ps后的原子快照。热浴过程中,钛原子在其初始位置附近并且不会移开,这表明在高温条件下TIG仍能保持结构的稳定性
附件下载:



